Sample Management Window
Sample Management window
Importing sequencing files and selecting samples
We advise you to store the sequencing data in a dedicated SBTengine analysis folder (e.g. SBTdata). If your computers are connected through a network we recommend you to store the sequence data on a file server that is accessible from each workstation where SBTengine is installed. Please make sure that you regularly make backups form this computer to prevent accidental loss of data.
Note: whether you use a networked environment or not, in this manual we will refer to the folder containing the sequencing files as the 'SBTdata' folder.
Operating procedure
- When the sequencing run is completed you need to copy the complete run folder from the sequencing computer to the SBTdata folder.
- Before ABI sequence files (i.e. *.ab1 files) can be analyzed by SBTengine, a sample and locus name must be defined to each ABI sequence file. This can be done in the Sample Management window in two ways: Automatic Name/Locus Assignment (recommended), Manual Name/Locus Assignment.
- SBTengine needs to know the location of the SBTdata folder. To do this, start SBTengine, go the Sample Management window and select the SBTdata folder in the left pane (1). Do NOT select a run folder, since SBTengine may combine data from several run folders. You only need to make this setting once per installation of SBTengine.
- Click the 'Hide Tree' button if you want to zoom in on all sequences in the specified SBTdata folder and hide the folder tree (2). This is particularly useful to prevent accidental deselection of your SBTdata folder.
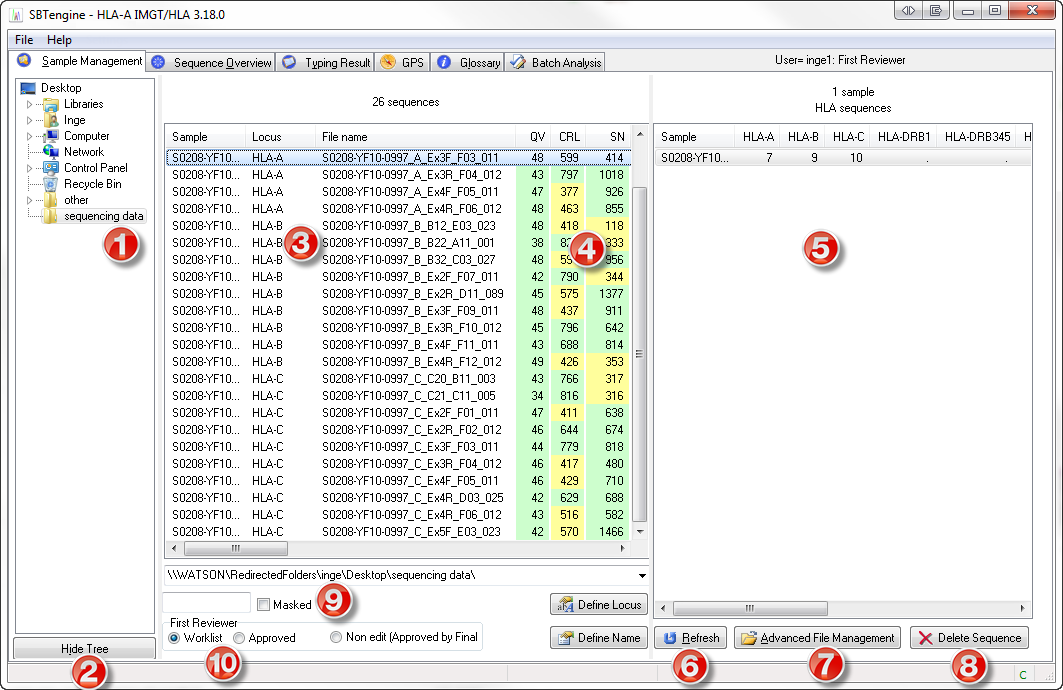
- All sequence files in the run folders that were copied to the SBTdata folder will appear in the middle pane (3). SBTengine registers all sequence files in the SBTdata folder and all subfolders. Sequence files located at a deeper level of the SBTdata folder are not detected.
- The middle pane also gives an overview of the sequence quality for each sequence trace (4), using a quality value (QV), contiguous read length (CRL) and signal noise ratio (SN). High QV, CRL and SN values are indicated in green, intermediate values in yellow, and low values in red. The color thresholds of these values can be adjusted in the preference settings. These quality values can also be used to generate quality report for approved samples in the Typing Result window.
- An overview of the sequence files is given in the right pane (5): the number of ABI sequence files per HLA locus is shown for each sample name.
- At the bottom of the right pane you can click the 'Refresh' button (6) to refresh the Sample Management window. This is particularly useful when new run folders are added while SBTengine is running. The list is automatically refreshed each time SBTengine starts up.
- Using the 'Advanced File Management' button (7) you may access the sequence files in the Windows Explorer.
- By clicking the 'Delete Sequence' button you delete the selected sequence files shown in the middle pane (8). This can be useful to remove sequence files of empty (water containing) wells in your sequencer plates.
- Checking the 'Masked' box (9) allows you to view a specific selection of the ABI sequence files based on the search string you enter. For example: entering 'Sample01' in the field before the 'Masked' box and pressing <Enter> will show you all ABI sequence files that contain the word 'Sample01' in any of the columns displayed in the middle pane.
- For explanation of the 'Worklist', 'Approved' and 'Non-edit' options (10), see Login and Reviewer levels.
How to add an additional sequencing file to an already analyzed and approved sample?
When a new sequencing run is completed, you can copy the new runfolder from the sequencer to the original SBTdata folder. When new sequencing information of a sample becomes available (e.g. after sequencing with GSSPs) this will be analyzed automatically together with data that was approved at an earlier stage. When a sample is selected within SBTengine, it will automatically look within the Archive map to detect if there is already data present. If so, these data will be opened again. The newly added data may change the final allele assignment, mostly by eliminating genotype or allele ambiguities.When the analysis is completed, the sample should be approved again. Newly added files will be moved to the archive map and a new XML file will be exported. SBTengine assumes that the latter allele assignment will be superior above the earlier one. Therefore the old XML file will be overwritten, without prior notice.
eDRB1 (feature only available for Abbott licenses)
The AlleleSEQR HLA-DRB1 use 3' amplification primers located in exon 2. This part in exon 2 may not be used for typing and is excluded from analysis in the EDRB1 database. In contrast, the DRB1 database contains the full sequence of exon 2. For correct analysis of DRB1 data generated with Abbott AlleleSEQR DRB1 kits, the sample file name should contain eDRB1 (or EDRB1). This will allow SBTengine to automatically select the correct database.
Example:
Correct naming: Sample1_eDRB1_Ex2Fa1.ab1
Incorrect naming (leading to use of the general DRB1 database): Sample1_DRB1_Ex2Fa1.ab1