This export can be generated using NGSengine 2.23 or higher. At the top of the file, you can find general information about the generated HML file.
These include the hml-id, reporting center-id and project. The <hmlid> is a code characterizing the generated file, which should not be changed.
The <reporting-center> contains the ‘Laboratory Code’ set in NGSengine.

For each sample, a section is added with the data. An HML export contains the data of a whole project, which means that there will be multiple sample sections.
Every sample has a sample id. That id is the one that can be found in the overview page of NGSengine. Center code, designates if the sample originates from the same lab that is generating the file or not. Finally, collection method is currently always reported to be unknown.

For each locus, the sample contains a "typing" section. It describes the gene family found, i.e HLA, the day an assignment was made to the locus of this sample, and information on the library used and its version.
The typing result is included as gl-string. The gl-string contains information which can also be found in the genotype ranking tab of NGSengine. The "typing method" section reports information concerning the source of the data, the NGS file format, and the actual file name(s), and some additional information.

Consensus sequence contains the "consensus" derived from the sequencer. That consensus is in relation to the sequence of a reference allele which most of time is the same as the best match saved in the gl-string. However, there are exceptions which cause a difference between the alleles reported in the gl-string and the consensus sequence source. These exceptions occur when the IPD-IMGT/HLA sequences of alleles reported in the gl-string are not covering the whole gene. In those cases NGSengine uses extrapolation. For the HML export the sequence of a fully sequenced allele must be used.
The following two sub parts are described in this section:
1) Reference sequence block
In this block we get the name of the reference allele to which the consensus-sequence-block refers to. For example, name=A*02:16 with the start and end position. Those values correspond with what is published in https://www.ebi.ac.uk/ipd/imgt/hla/allele.html.
2) Consensus sequence blocks
These blocks describe the sequences from the phasing regions with respect to the reference sequences, also reporting with which reference, and which region in the reference it matches.
Within the consensus sequence block we find:
- reference-sequence-id: the first exported allele of each project is ref1, the second ref 2, etc.
- start/end: start or end position of consensus sequence compared to reference sequence, start is always "0" and end is the length of allele published in the IMGT/HLA database
- strand: always -1
- phase-set: describing the phased region this sequence belongs to
- continuity: true or false > true if the next consensus sequence block starts immediately after the previous (next nucleotide), false if there is an ignored region after for example
- expected-copy-number: always 1
Example of two alleles with a perfect match in the IMGT/HLA database:

The HML format indicates the sequence found in a sample by means of comparing with a reference allele from the IMGT/HLA database. Any differences in sequence are reported through variant elements. These blocks describe the position of the reference where the variant starts and stops, the original nucleotides of the reference and the actual nucleotides of the allele in the sample.
Example of a new allele with a single nucleotide change, a change from "G" to "C":
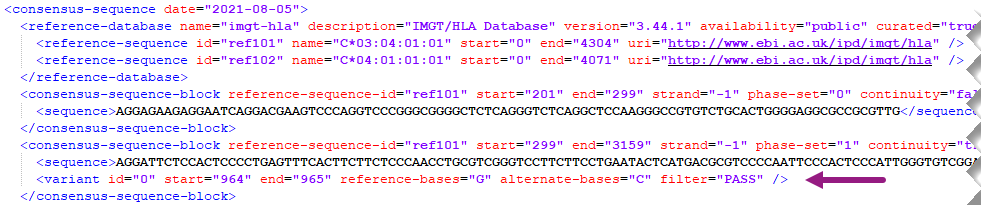
Example of a single nucleotide deletion, a change from "CG" to "C":

A few more examples:
1) Only one variant created:
Reference sequence: ACTGG
Actual sequence: ACAAA
reference-bases = "TGG" alternate-bases = "AAA"
2) Two variants created:
Reference sequence: ACTGG
Actual sequence: ACAGA
reference-bases = "T" reference-bases = "G"
alternate-bases = "A" alternate-bases = "A"
3) Variant with deletion of 2 nucleotides:
Reference sequence: AAATT
Actual sequence: AA . .T
reference-bases = "AAT" alternate-bases = "A"
- Make sure that all loci within the project have been analyzed with the same IMGT/HLA library version