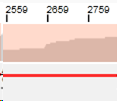
Coverage bar

The numbers on top indicate the position in the gene. The numbering presented here is identical to the IMGT genomic numbering.
The numbering can be switched to cDNA or codon numbering, as described here.
The coverage bar shows multiple vertical bars. Each bar represents the read depth (the number of reads on each position) on a specific position.
A grey bar indicates that the data is homozygous at this specific position.
Colored bars indicate positions where NGSengine has determined the data to be heterozygous (heights are proportional to the ratio of the heterozygous base calls).
By hovering the mouse over these bars, a pop-up screen appears, showing the information about this specific position.
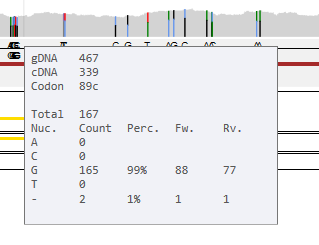
This pop-up screen shows from top to bottom:
- In a coding region: gDNA position, cDNA position and codon position. In an intron: only the gDNA position.
- Total number of reads covering this position
- Nucleotide counts for A, C, G, T or deletions (-) found at this position. Between brackets the percentages of these base calls and the number of forward reads (+) and reverse reads (-).
By clicking on a certain position in the coverage bar, NGSengine will zoom in on that position.
When a part of the coverage bar is colored pink, this indicates that the read depth in this part is lower than the threshold. As a default, this threshold is set to 20 reads. This part is excluded from analysis. The read depth threshold can be changed via the preferences and subsequent re-analysis of this locus.