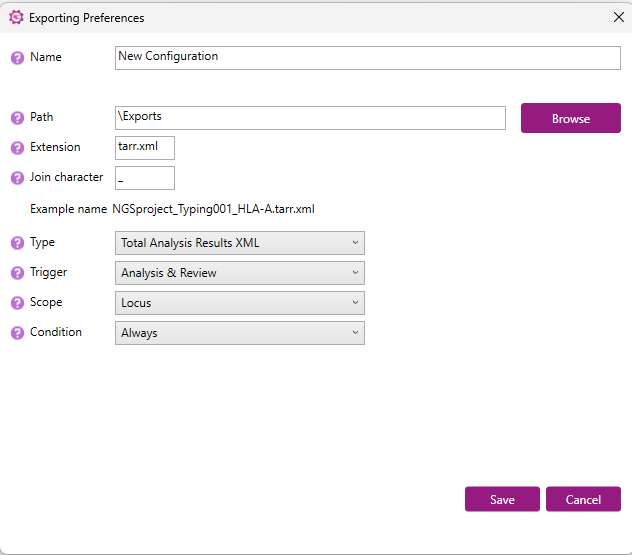
Exporting
The Total Analysis Results export can be used to generate XML files.
The tarr.xml starts with information about locus name, sample name and user name. This is followed by read information, question mark positions, phasing region, number of genotypes and IMGT version.
Next, the XML tab is divided in a number of sections:
Regions
This section describes the start and end positions of the exons, introns, and UTRs. Position numbers match the genomic numbering of the IMGT/HLA website.
Priority Regions
This section describes the regions that have priority for genotype analysis. The priority regions include all exons and their splice sites, but also certain parts of the introns and UTR that are important for the correct identification of particular low expressed alleles and null alleles.
Haplotypes
This section describes the areas of the sequence that can belong to either allele 1, allele 2, or both alleles. The nucleotide sequences of all homozygous and heterozygous areas are described here, together with the first and last position. For each heterozygous area, two nucleotide sequences are reported with suffix ‘.1’ and ‘.2’, representing the two alleles.
The homozygous and heterozygous areas can be combined to form the whole sequence of both alleles. Which of the areas can be combined to form the best match, is described in the next section (Matches).
Matches
This section describes how the sequences of the homozygous and heterozygous areas should be combined to form the best matching genotype.
For example, in case a sample shows two heterozygous areas (2 phasing regions), three homozygous areas will be reported as well: one homozygous area in between the two heterozygous areas and 2 homozygous areas flanking both sides.
Homozygous1 represent both alleles at the first homozygous region.
Heterozygous1.1 and heterozygous1.2 represent the two alleles at the first heterozygous region.
An allele matching with phasing = “1,1” has sequence:
homozygous1 + heterozygous1.1 + homozygous2 + heterozygous2.1 + homozygous3.
In this configuration, the second allele is represented by phasing= “2,2”:
homozygous1 + heterozygous1.2 + homozygous2 + heterozygous2.2 + homozygous3.
Match Sets
This section describes how many mismatches there are in the introns, exons, priority regions, and non-priority regions. For each allele within the match, the phasing, number of mismatches, and the location and content of the mismatches are listed. This section also describes for each mismatch which number in the sequence is mismatched, and to which IMGT/HLA database number this corresponds.
Typing result
This section shows a list of the genotypes with 0 Exon+ mismatches, including the CWD, P group, G group and Serology information.
The XML tab ends with information about the used software version and the analysis settings.