Settings for Pacbio data
NGSengine handles two types of PacBio data:
- Subreads (also called raw reads)
- Consensus reads: these are pre-analyzed by the PacBio Long Amplicon Analysis tool (LAA) and contain only a small number of reads (one or two reads per gene)
Subreads:
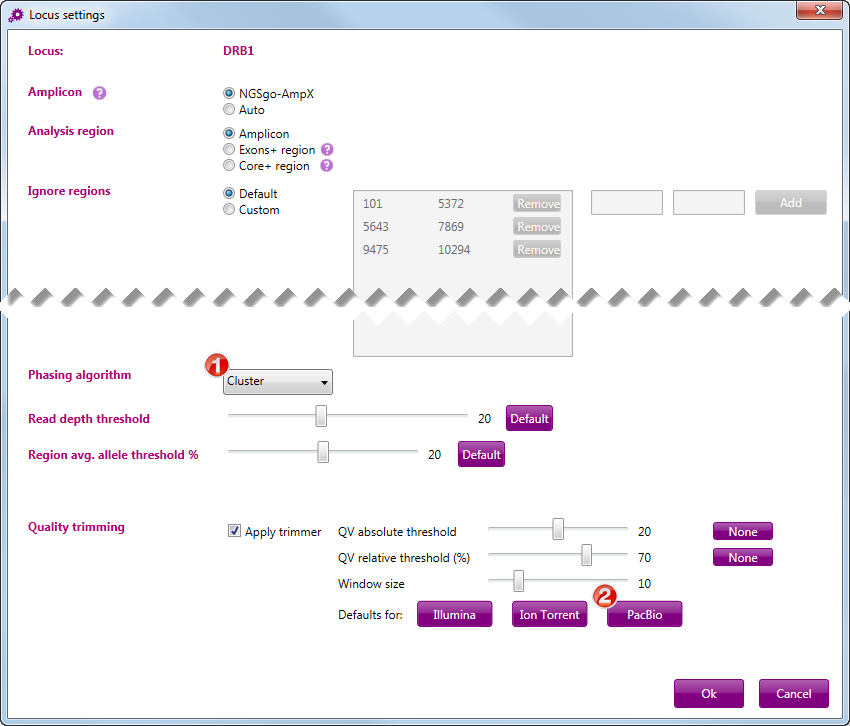
Go to File > Preferences > Locus default settings.
For each locus separately, select the following settings:
- Phasing algorithm: Cluster (1)
- Quality trimming: select the default settings for PacBio (2)
NGSengine recognizes if these files contains reads >1250 bp. These are analyzed automatically in PacBio mode.
Typical results with subreads:
- Noise levels ~10%
- Lower mappability per gene (5-30%) caused by higher noise levels, sequence artefacts and very long reads
- Relatively many single base insertions and deletions, homopolymer issues, potentially hampering phasing
- Single basepair artefacts and homopolymer issues can be avoided by excluding these positions from analysis
Consensus reads:
Check that the cluster phaser is applied (see picture above), for each locus separately.
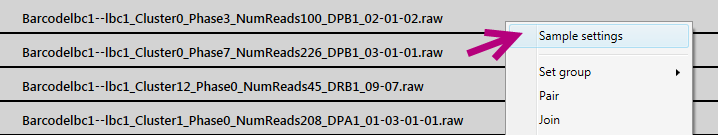
Switch on PacBio consensus mode by selecting sample(s) > click right mouse button > Sample settings:
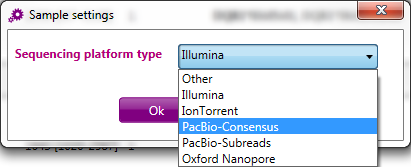
A window opens > select PacBio consensus > click OK:
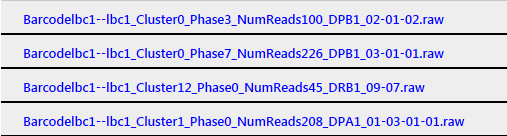
Sample name turns blue:
Other classifier, aligner and phaser will be applied, which are optimized for low number of long reads.
Typical results with consensus reads:
- Mappability in most cases 100%