Reads
Initial view:
In this part of the screen, individual reads can be presented. By default, the reads are not shown.
The reads can be made visible by clicking the 'Show Reads' button in the lower right corner:
![]()
Analysis screen with all reads displayed:
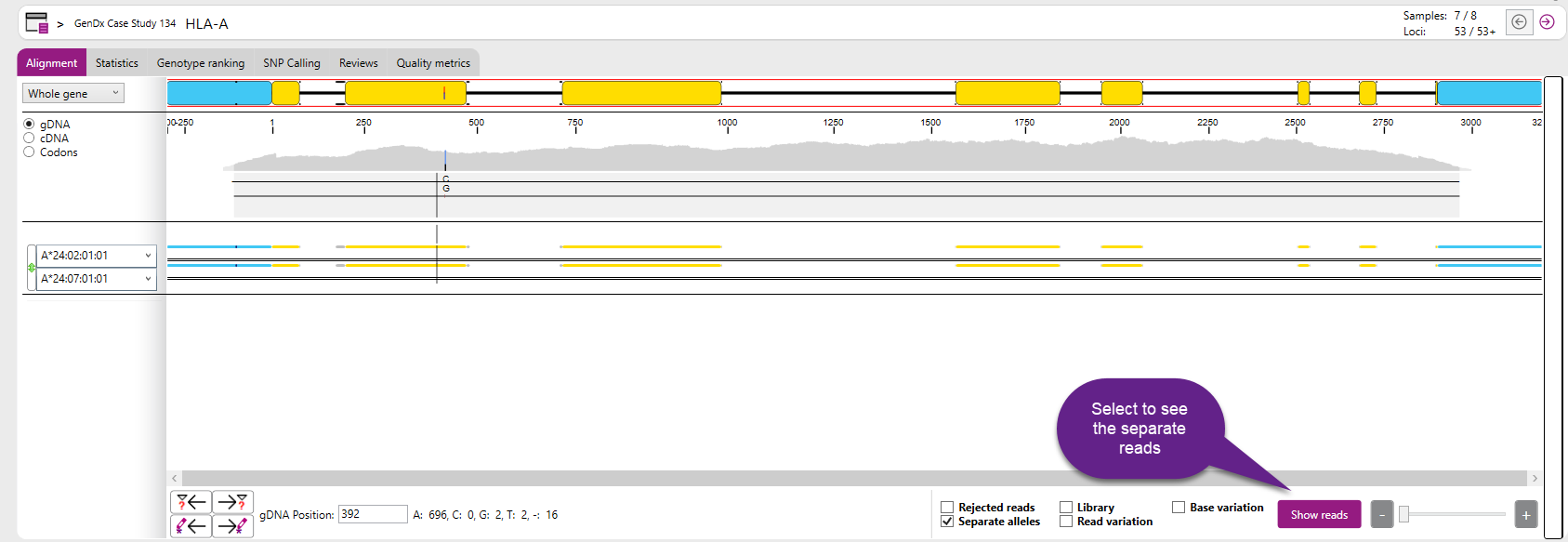
To hide the reads of both alleles, click the "Hide reads" button.

Via the 'View' option in the menu bar, as well as via the checkboxes at the bottom of the screen, different options regarding organization of the reads can be chosen.
When settings are changed via the view menu, they will be remembered and used by default. This 'View' option is further explained here.
Settings changed in the lower part of the overview screen will be applied for this locus only. These buttons are explained in more detail here.

Each grey bar represents a single read. Everywhere where the line is grey, the sequence is identical to the reference. The green, red, black and blue colors, indicate positions where the sequence is different from the reference.
After zooming in, more details become visible:
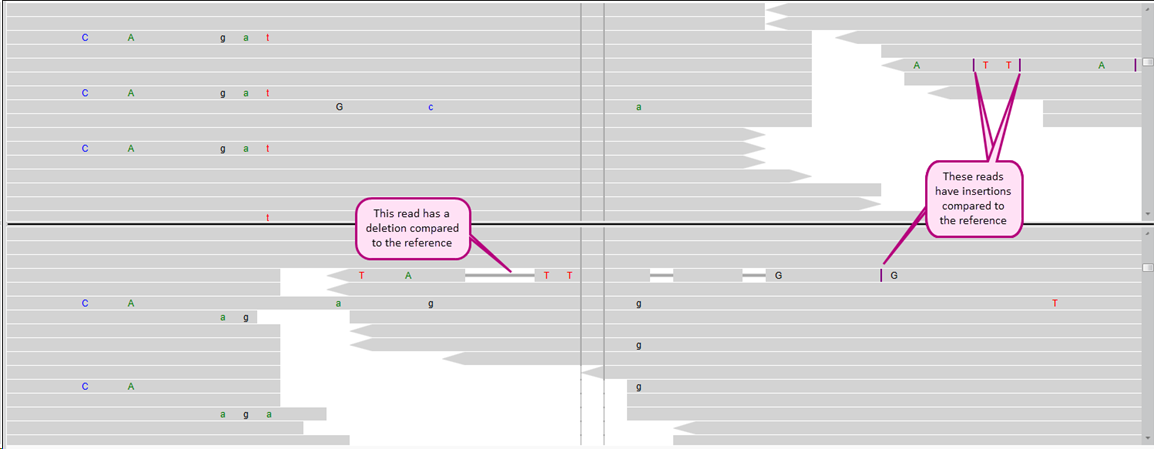
Arrows at the end or beginning of reads indicate if the read has the forward or reverse direction.
Thinner grey horizontal lines indicate a deletion in the read in comparison to the reference(s).
Vertical purple lines indicate an insertion in the read in comparison to the reference(s).
Capital letters represent nucleotides in the read which differ from both of the two references.
Small letters represent nucleotides in the read which differ from the allele to which the read was aligned, but are identical to the other reference allele.
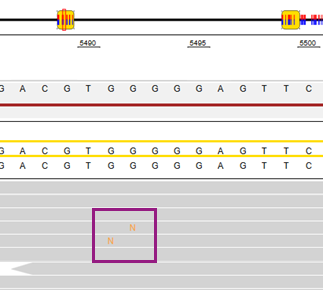
Orange Ns can be shown in the reads in the alignment view. This occurs when the sequencer assigns a low quality call to that position, meaning it could be any base (A, C, T, or G).
When hovering the mouse over the reads, more information concerning this specific read and position is shown:
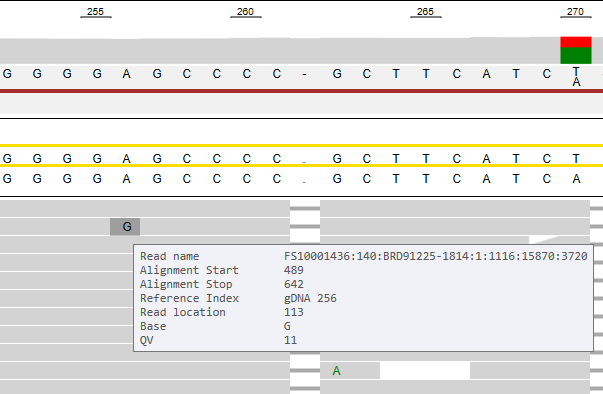
When hovering to the left of an insertion (vertical purple line), the pop-up window will also present which nucleotide is inserted.
By right-clicking on a specific nucleotide of a read, additional options appear:
Sort on position
Sorting on a specific position will rearrange the reads in such a way that identical nucleotides will be grouped together in the alignment.
Before sorting on position:
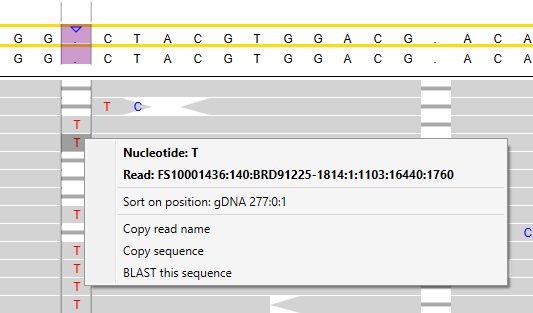
After sorting on position:

After sorting on a specific position, all A, C, G, and T nucleotides at this position have been grouped together. In the image above, you can see the T nucleotides grouped together.
Copy read name:
The read name will be copied to the clipboard.
Copy sequence:
The nucleotide sequence of the read will be copied to the clipboard, this is the trimmed sequence when trimming has been applied.
It is the sequence as determined by the sequencer, so for reverse reads it is the reverse compliment of what you see on the alignment screen.
BLAST this sequence
The nucleotide sequence of the read you selected will be copied to NCBI BLAST. Your internet browser will open and BLASTing starts immediately.
This function can be useful when patterns are found that do not seem to belong to either allele in the sample. BLASTing can identify for example whether a read originates from a different locus or a pseudogene.
More information about BLAST can be found on the NCBI website.