View options
At the top there is a drop-down menu which enables viewing of different regions:
- Whole gene: the complete gene is shown.
- Amplicon: only the amplicon is shown.
- Exon+: only the exons plus flanking priority regions (including all splice sites) are shown. Priority regions are intron regions that enable correct identification of the presence or absence of particular null alleles (includes all splice sites).
- Core+: Only the core regions plus flanking priority regions are shown. The Core+ region(s) are exon 2 and 3 for class I and exon 2 for class II genes, including flanking priority regions.
When the view is changed in the alignment window, the same view is applied in the statistics view.
At the bottom of the analysis screen there are several buttons present:
![]()
At the right there is the 'Show reads'/ 'Hide reads' button: when clicking this button all reads used for the analysis of both alleles are being displayed or hidden.
There are 3 ways to zoom in or out of the reads:
- Click on the + and – buttons in the zoom bar
- Drag the cursor in between the + and – buttons
- Hold the CTRL key on your keyboard in combination with the mouse wheel
At the bottom left of the analysis screen there is the possibility to Jump to a specific gDNA position. Just enter the position and press enter and NGSengine jumps to the requested position.

At the bottom right of the analysis screen there are several checkboxes:

The information selected by each checkbox is displayed in the following order:
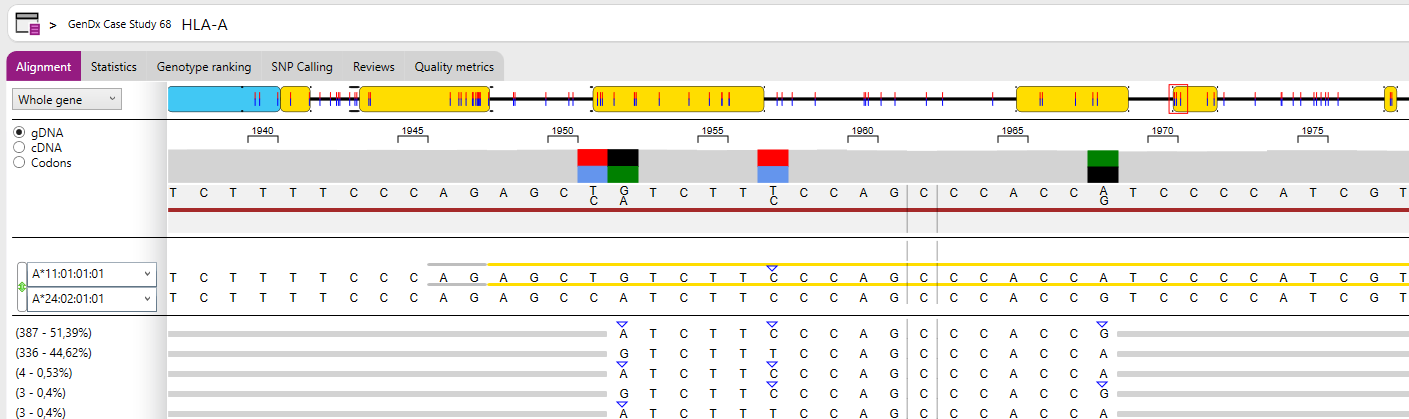
- Base variation: The Base variation plot is displayed below the reference alleles.
- Separate alleles: The reads of allele 1 and allele 2 will be shown separated by a black line.
- Rejected reads: Shows the rejected reads which are excluded from the analysis.
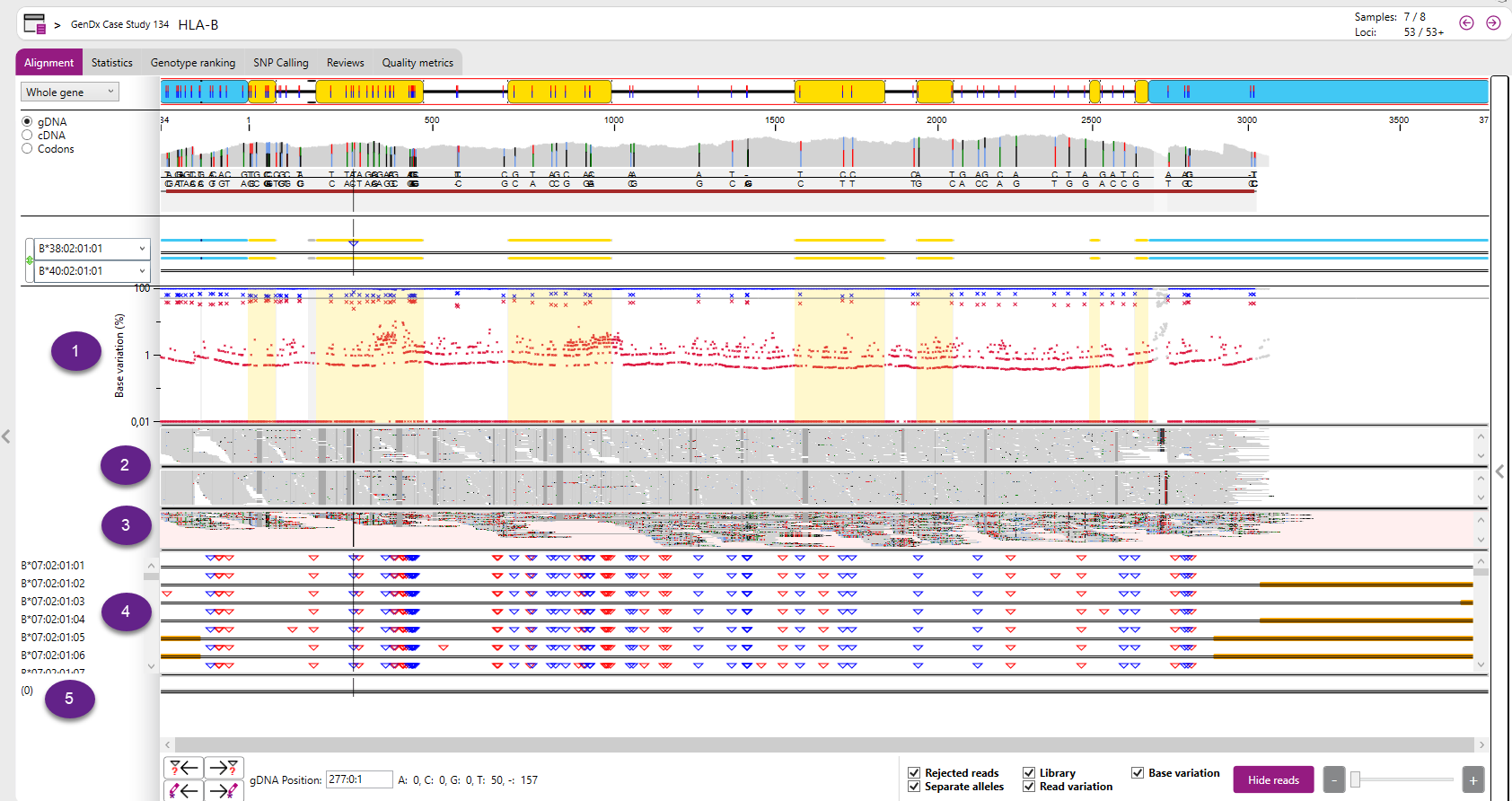
- Library: Shows all sequences in the database for the selected positions and compares them to the first reference allele. The number between (parenthesis) indicates how many alleles have the same sequence for the selected positions. You can select a single position by clicking on the position of interest. You can select multiple positions by clicking and dragging, as in the view below.
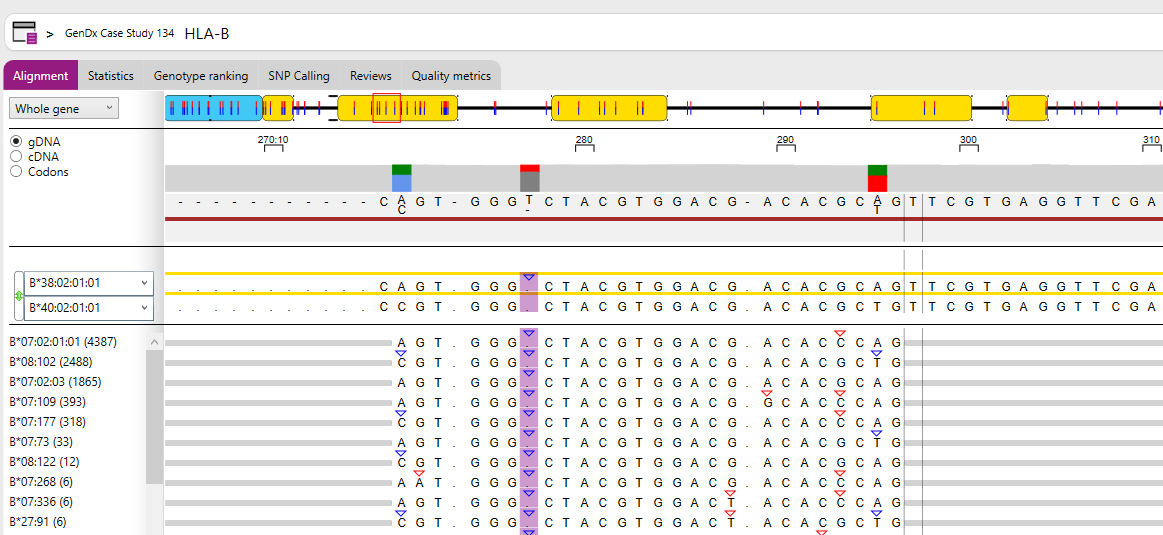
- Read variation: shows all patterns found in the reads for the selected positions and compares them to the first reference allele. You can select a single position by clicking on the position of interest. You can select multiple positions by clicking and dragging, as in the view below. The numbers between (parenthesis) indicate how many reads were found with each pattern in the data, and the percentage based on the total of reads that cover the selected region.