Pacbio consensus reads
When Pacbio consensus reads are used for analysis, the reads will be shown differently in the alignment screen then for other data. The reads are larger, the name of the read is shown above each read and the reads are numbered (1, 2, 3, etc.):

The reads that are shown in dark grey have been used for the typing. The reads that are shown in light grey have been aligned but not used for typing.
Below the reads a table is shown in which the aligned reads are compared to each other for mismatch level:
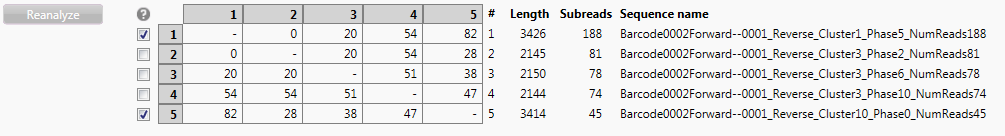
The reads used for typing are checked in the checkboxes next to the table. Behind the table is shown for each read: the length of the read, the number of subreads that have been used to create the consensus read and the sequence name.
It is possible to include other reads in the analysis, by checking or unchecking the checkboxes in the table. By (de)selecting reads, the 'Reanalyze' button will become activated:
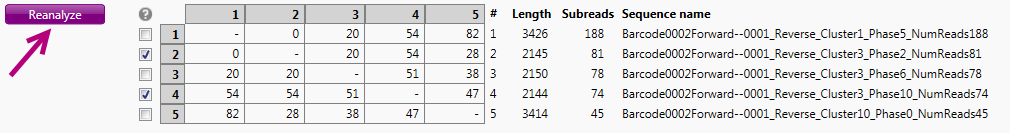
By clicking this button, the reads selected will be used for a new analysis.
In principle a maximum of 2 reads can be used for analysis, 1 read per allele. The only exception is when there are 2 reads for an allele that do not overlap in the alignment, then both reads can be selected for analysis which will result in a total of 3 reads (2 reads for allele 1 and 1 read for allele 2) or 4 reads (2 reads for each allele) whcih can be used for analysis.